An open-source pipeline to construct a global phylogeny of the plague pathogen Yersinia pestis.
![]()
![]()
![]()



Pipeline Overview
- Create a metadata database of NCBI genomic assemblies and SRA data (
NCBImeta) - Download assemblies and SRA fastq files (
sra-tools) - Build SnpEff database from reference (
SnpEff) - Align to reference genome (
snippy,eager) - Mask problematic regions (
dustmasker,mummer,vcftools) - Evaluate statistics (
qualimap,multiqc) - Construct a Maximum Likelihood phylogeny (
iqtree) - Optimize time-scaled phylogeny (
augur,treetime) - Web-based narrative visualization (
auspice)
Showcase
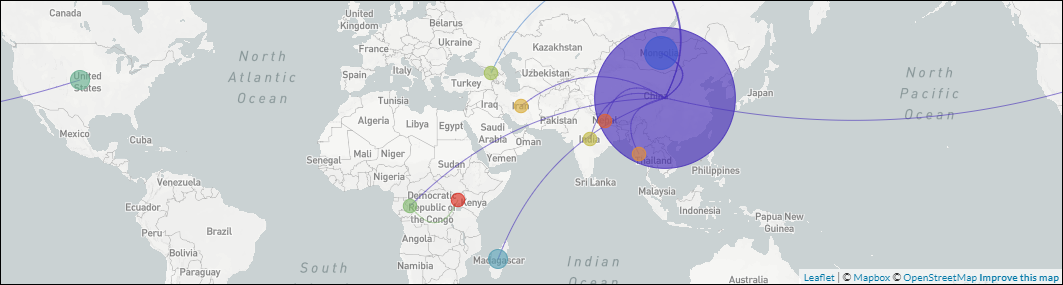
- Presenting “The Plague”: Digital Exhibits as Interdisciplinary Method.
DHSI Conference and Colloquium. June 5, 2020.
Katherine Eaton, Nukhet Varlik, Ann Carmichael, Brian Golding, Hendrik Poinar.
Digital Exhibit • Talk
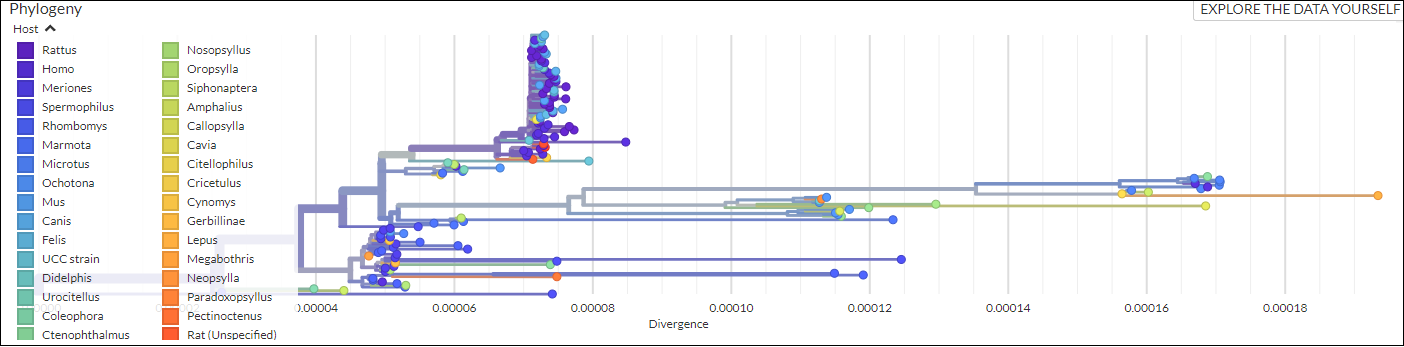
- Plagues of the Past and Present.
Lewis & Ruth Sherman Centre for Digital Scholarship. June 2, 2020.
Katherine Eaton
Digital Exhibit • Blog Post 1 • Blog Post 2 *
Install
All install options start by cloning the pipeline repo.
git clone https://github.com/ktmeaton/plague-phylogeography.git
cd plague-phylogeography
1. Conda (Laptop)
conda install -c conda-forge mamba
mamba env create -f workflow/envs/merge/environment.yaml
conda activate plague-phylogeography
snakemake --profile profiles/laptop help
(While mamba is not strictly necessary, it is heavily recommended.)
2. Docker (Laptop)
docker pull ktmeaton/plague-phylogeography:dev
docker run \
-v $PWD:/pipeline \
-w /pipeline \
ktmeaton/plague-phylogeography:dev \
snakemake --profile profiles/laptop help
3. Singularity (HPC - Compute Canada)
singularity pull docker://docker.io/ktmeaton/plague-phylogeography:dev
singularity exec plague_phylogeography_dev.sif \
snakemake --profile profiles/compute-canada help
If you will be downloading data from the SRA with singularity, the sra toolkit must be configured:
mkdir -p ~/.ncbi/
printf '/LIBS/GUID = "%s"\n' `uuidgen` > ~/.ncbi/user-settings.mkfg;
Credits
Author: Katherine Eaton
Logo: Emil Karpinski, Katherine Eaton