Katherine Eaton 0000-0001-6862-7756
· ktmeaton
Ancient DNA Centre, McMaster University; Department of Anthropology, McMaster University
· Funded by Social Sciences and Humanities Research Council XXXXXXXX
Leo Featherstone 0000-0002-8878-1758
The Peter Doherty Institute For Infection and Immunity, University of Melbourne
Hendrik Poinar 0000-0002-0314-4160
Ancient DNA Centre, McMaster University; Department of Anthropology, McMaster University
Abstract
Introduction
Plague has an impressively long and expansive history as a human disease. The earliest evidence of the plague bacterium, Yersinia pestis, comes from ancient DNA studies, dating its emergence to at least the Neolithic [1,2]. Since then, Y. pestis has traveled extensively due to ever-expanding global trade networks and the ability to infect a wide variety of mammalian hosts [3,4]. Few regions of the ancient and modern world remain untouched by this disease, as plague has an established presence on every continent except Oceania [5].
Accompanying this prolific global presence is unnervingly high mortality. The infamous medieval Black Death is estimated to have killed more than half of Europe’s population [6]. This virulence can still be observed in the post-antibiotic era, where case fatality rates range from 22-71% [7]. As a result, plague maintains its status as a disease that is of vital importance to current public health initiatives.
This high priority disease status is unsurprising given that Y. pestis is a member of the Enterobacteriaceae family. This family includes other notable pathogens such as Escherichia coli and Salmonella typhi that are commonly transmitted by contaminated food and water. In comparison, the plague bacterium is unique among this family due to a striking difference in host habitat and transmission. Y. pestis commonly resides in the blood of its mammalian hosts and can be transmitted to new hosts through an infectious fleabite [8]. Furthermore, this unique mechanism evolved relatively recently, possibly around the 1st millennium BC [9], long after Y. pestis emerged as a monomorphic clone of the enteric pathogen Yersinia pseudotuberculosis[10].
While the population structure of Y. pseudotuberculosis is well-defined, [11,12], the phylogenetic patterning of Y. pestis remains cryptic. Populations of Y. pestis have been historically categorized according to a a vast array of historical, ecological, biochemical, and molecular characteristics. As a result, disparate sub-typing systems have emerged over the years to differentiate lineages of plague [13]. It has thus been argued that the taxonomy of Y. pestis should be revised and consolidated according to the latest global phylogenetic analysis [14].
Unfortunately, there are a number of obstacles that have stalled large-scale phylogenetic analysis. The first challenge is data availability, both in terms of the genomic sequences, as well as the metadata required for interpretation. Genomic sampling of Y. pestis has recently intensified
[15], thus providing exceptional new datasets for statistical inference. This intensive sampling has produced over 1000 Y. pestis genomes that are now publicly available, with tremendous diversity spanning five continents and 5000 years of human history. Unfortunately, the majority of these genomic records lack curated metadata, such as sampling date and location, which are crucial variables in testing population structure.
The second major obstacle is an apparent lack of temporal structure in Y. pestis. Detecting temporal signal and estimating a molecular clock model are general pre-requisites for sophisticated methods of quantifying population structure [16].
However, there has been significant debate concerning whether Y. pestis can be appropriately modeled using the available clock methods [17,18,19].
To some extent, this debate can be explained by different Y. pestis datasets, which have been shown to produce dramatically different patterns of temporal signal [20]. Therefore, it is uncertain whether the new intensively sampled genomes will bring clarity or greater uncertainty.
In response to these debates and obstacles, this paper proposes a theoretical and methodological shift in plague genomics. Rather than conceptualizing Y. pestis as a conglomerate species, we highlight how novel insight emerges through analyzing Y. pestis sub-populations in isolation. To accomplish this shift in discourse, we focus on four objectives, specifically to:
Curate and contextualize the most recent Y. pestis genomic metadata.
Review and critique our current understanding of Y. pestis population structure.
Conduct robust and nuanced molecular clock analyses.
Identify key areas of phylogenetic uncertainty to be expanded on in future research.
Progress towards these key objectives is anticipated to benefit both prospective studies of plague, such as environmental surveillance and outbreak monitoring, and retrospective studies, which seek to date emergence and spread of past pandemics.
Methods
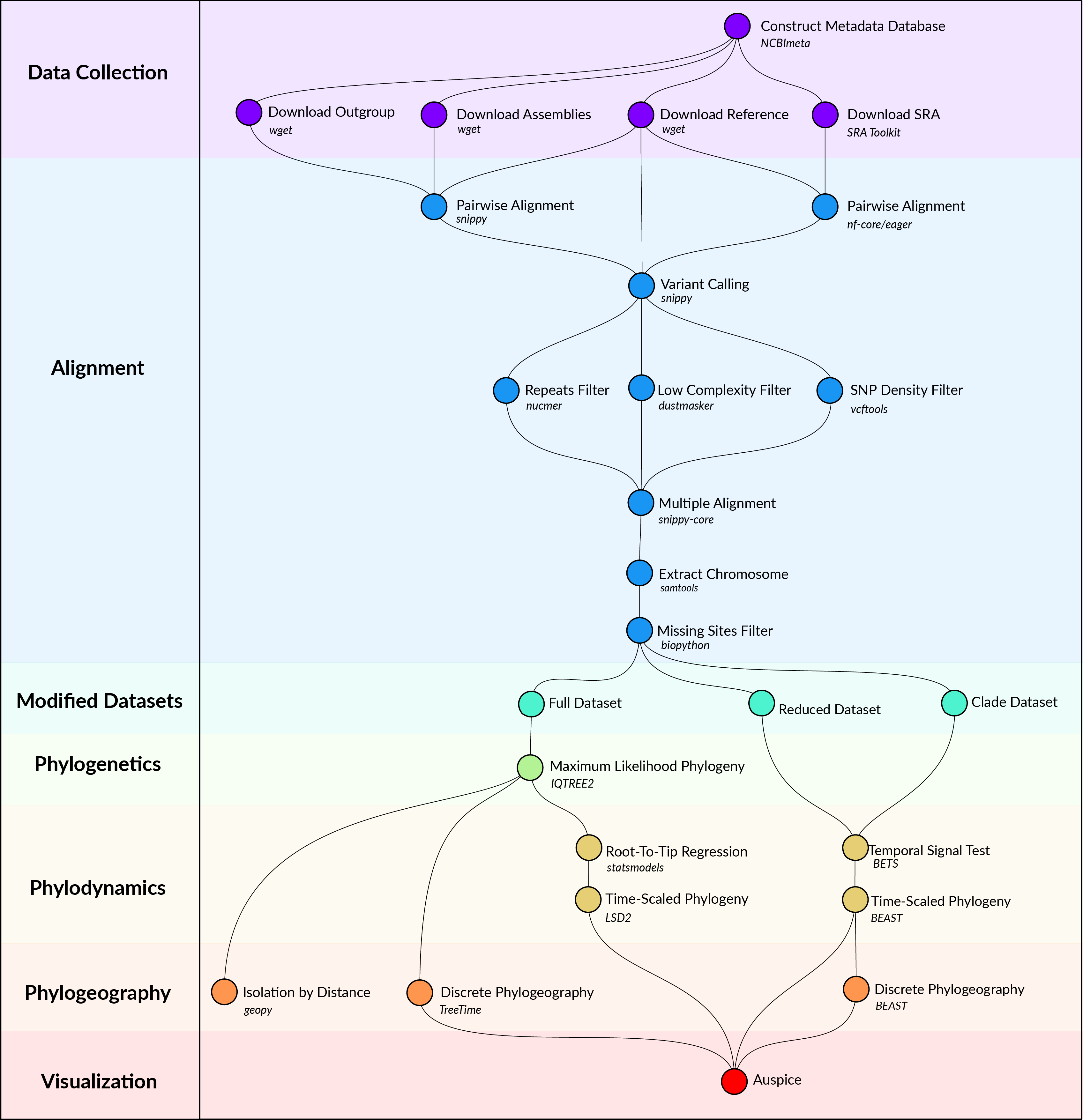
A visual overview of the computational methods is provided in Figure 1 and is available as a snakemake pipeline (https://github.com/ktmeaton/plague-phylogeography/).
Figure 1: Computational methods workflow.
Data Collection
Y. pestis genome sequencing projects were retrieved from the NCBI databases using NCBImeta [21]. 1657 projects were identified and comprised three genomic types:
586 modern assembled
184 ancient unassembled
887 modern unassembled
The 887 modern unassembled genomes were excluded from this project, as the wide variety of laboratory methods and sequencing strategies precluded a standardized workflow. In contrast, the 184 ancient unassembled genomes were retained given the relatively standardized, albeit specialized, laboratory procedures required to process ancient tissues. Future work will investigate computationally efficient methods for integrating more diverse datasets.
Collection location, collection date, and collection host metadata were curated by cross-referencing the original publications. Collection location was transformed to latitude and longitude coordinates using GeoPy [22] and the Nominatim API [23] for OpenStreetMap [24]. Coordinates were standardized at a sub-country resolution, taking the centroid of the parent province/state. Collection dates were standardized according to their year, and recording uncertainty arising from missing data and radiocarbon estimates. Collection host was the most diverse field with regards to precision, ranging from colloquial nomenclature (“rat”) to a genus species taxonomy (“Meriones libycus”). For the purposes of this study, collection host was recorded as Human, Non-Human, or Not Available, given the inability to differentiate non-human mammalian hosts.
Genomes were removed if no associated date or location information could be identified in the literature, or if there was documented evidence of laboratory manipulation.
Two additional datasets were required for downstream analyses. First, Y. pestis strain CO92 (GCA_000009065.1) was used as the reference genome for sequence alignment and annotation. Second, Yersinia pseudotuberculosis strains NCTC10275 (GCA_900637475.1) and IP32953 (GCA_000834295.1) served as an outgroup to root the maximum likelihood phylogeny.
Alignment
Modern assembled genomes were aligned to the reference genome using Snippy, a pipeline for core genome alignments [25]. Modern genomes were removed if the number of sites covered at a minimum depth of 10X was less than 70% of the reference genome.
Ancient unassembled genomes were downloaded from the SRA database in FASTQ format using the SRA Toolkit [26]. Pre-processing and alignment to the reference genome was performed using the nf-core/eager pipeline, a reproducible workflow for ancient genome reconstruction [27]. Ancient genomes were removed if the number of sites covered at a minimum depth of 3X was less than 70% of the reference genome. It is a typical approach to relax coverage thresholds for ancient genomes relative to their modern counterparts [cite?]. The threshold chosen here is commonly used, and aims to strike a balance between variant confidence and sample representation [cite?].
A multiple sequence alignment was constructed using the Snippy Core module of the Snippy pipeline [25]. The output alignment was filtered to only include chromosomal variants and to exclude sites that had more than 5% missing data.
Modified Datasets
To investigate the influence of between-clade variation in substitution rates, the multiple alignment was separated into the major clades of Y. pestis, which is referred to as the clade dataset. Clade labeling was derived from the five-branch population structure accompanied by a biovar abbreviation [17]. Only one modification was made, in that the subclade associated with the Plague of Justinian (0.ANT4) was considered to be a distinct clade from its parent (0.ANT) due to its geographic, temporal, and ecological uniqueness.
To improve the performance and convergence of Bayesian analysis, a subsampled dataset was constructed and is referred to as the reduced dataset. Clades that contained multiple samples drawn from the same geographic location and the same time period were reduced to one representative sample. The sample with the shortest terminal branch length was prioritized, to diminish the influence of uniquely derived mutations on the estimated substitution rate. An interval of 25 years was identified as striking an optimal balance, resulting in 191 representative samples.
Phylogenetics
Model selection was performed using Modelfinder which identified the K3Pu+F+I model as the optimal choice based on the Bayesian Information Criterion (BIC) [28]. A maximum-likelihood phylogeny was then estimated across 10 independent runs of IQTREE [29]. Branch support was evaluated using 1000 iterations of the ultrafast bootstrap approximation, with a threshold of 95% required for strong support [30].
Phylodynamics
To explore the degree of temporal signal present in the data, two categories of tests were performed . The first was a root-to-tip (RTT) regression on collection date using the python package statsmodels[31]. Given the relative simplicity of a regression model, the full dataset of 601 genomes was used. For the second test of temporal signal, a Bayesian Evaluation of Temporal Signal (BETS) was conducted. As the complexity of Bayesian modeling is computationally intensive, the reduced dataset (N=191) was used.
Kat’s Notes:
- I will need Sebastian and Leo’s input here to write the BEAST methods section.
Public Resource
The maximum likelihood and Bayesian phylogenetic trees were uploaded to the Nextstrain visualization platform at https://nextstrain.org/community/ktmeaton/plague-phylogeography-projects@main. All curated metadata is available for download via Nextstrain, Github, and Zenodo.
Kat’s Notes:
- I’ll need to prepare stable and archived hosting links before submission.
Results and Discussion
Phylogeny
A maximum-likelihood phylogeny was estimated from 603 genomes (600 Y. pestis isolates, 1 Y. pestis reference, and 2 Y. pseudotuberculosis outgroup taxa). Following removal of the outgroup taxa, the alignment was composed of 10,249 variant positions with 3,844 sites shared by at least two genomes. In Figure 2, the maximum-likeilhood phylogeny is visualized alongside the four major taxonomic systems currently used to define the population structure of Y. pestis. These include the major phylogenetic branches, biovars, subspecies, and historical time periods/pandemics.
Figure 2: The maximum-likelihood phylogeny depicts the global population structure of Y. pestis. The divisions of the four major sub-typing systems are provided.
Population Structure
A comparison of sub-typing systems reveals great uncertainty with regards to Y. pestis population structure. The global phylogeny of Y. pestis can be divided into major branches according to the relative position of the “big bang” polytomy [17]. All lineages that diverged prior to this multifurcation are grouped into Branch 0 and those emerging after are the monophyletic clades Branches 1-4. Because the “big bang” polytomy plays such a central role in this system, there is growing interest in estimating its timing and geographic origins [32]. However, an inability to identify phenotypes distinguishing these major branches poses a significant challenge, and thus the exact epidemiological significance of the “big bang” polytomy remains unclear.
An example of this challenge can be seen in the population structure defined by the biovar system. Y. pestis can be categorized using a suite of metabolic properties into the classical biovars: antiqua (ANT), medievalis (MED), orientalis (ORI), and microtus/pestoides (PE) [33,34,35]. While these divisions don’t fully contradict the major branches, they do considerably shift the defining boundaries between Y. pestis populations.
To further complicate matters, researchers from the Commonwealth of Independent State (CIS) have observed biovar inconsistencies in plague foci surveillance [14]. In response, the subspecies taxonomy was created to distinguish a main subspecies (pestis) that is highly virulent in humans with wide geographic spread, from five or more non-main subspecies that have limited geographic ranges and attenuated virulence in humans [14]. The subspecies structure is highly convenient for laboratory diagnostics in the CIS, but struggles to differentiate the immense diversity represented by the larger pestis subspecies.
The challenge of categorizing plague by metabolism is, unsurprisingly, also an obstacle when analyzing extinct lineages. Ancient DNA (aDNA) researchers have opted to either extrapolate an existing biovar designation [18] or create a new one [9]. However, it is more common in aDNA studies to define population structure by time period and associations with historically documented pandemics. The known genetic diversity of Y. pestis thus far has been broadly grouped into four time periods and three pandemics (Figure 3).
Figure 3: The temporal distribution of Y. pestis genomes.
The First Pandemic (6th - 8th century CE) began with the Plague of Justinian and proceeded to devastate the Byzantine Empire of the Mediterranean world [cite?]. The Late Antiquity clade found within Branch 0 (0.ANT4) is associated with this pandemic given spatiotemporal overlap of the skeletal remains from which this lineage was retrieved [18,36]. Similarly, the medieval clade 1.PRE from Branch 1 is thought to derive from the Second Pandemic of Plague. This well-documented pandemic began with the infamous Black Death and swept across most of Eurasia from the 14th to 19th centuries [cite?]. The third documented pandemic of plague, alias the Modern Pandemic, spread globally from the end of the 18th Century and until the mid-20th Century. There is little dispute that a new lineage of plague emerging from Branch 1 as biovar orientalis (1.ORI) was the causative agent of this pandemic. While the World Health Organization (WHO) declared the third pandemic over in 1950 [cite?], this lineage continues to re-emerge to cause localized epidemics such as the 2010 plague in Peru [cite?] and the Madagascar Outbreaks of 2017 [cite?].
While the pandemic clade nomenclature provides an excellent foundation for historical discussion, there are several emergent problems with this system. First is the growing awareness of the spatiotemporal overlap of the Second and the Third Pandemic [cite?]. Previously, the temporal extents of these events were mutually exclusive, dating from the 14th-18th century, and the 19th-20th century respectively. Recent historical scholarship has contested this claim, and demonstrated that these constraints are a product of a Eurocentric view of plague [cite?]. The Second Pandemic is now known to have extended into the 19th Century in parts of the Ottoman Empire, with the latest epidemics dating to 1819 [cite?]. Similarly, the Third Pandemic is now hypothesized to have began as early as 1772 in southern China [37]. It remains unclear where to draw the distinction, if it even exists, between the Second and Third Pandemic.
Another limitation of the pandemic nomenclature is the complete disconnection of Branch 2 to any historical pandemics. This is surprising given that several criteria of a pandemic pathogen are fulfilled by Branch 2 lineages, namely extensive spread and virulence. Branch 2 genomes of Y. pestis have been collected from all throughout Eurasia, stretching from at least the Caucasus, to India, and to eastern China (Figure 4). Furthermore, lineages of Branch 2 have been associated with high mortality epidemics [38] and were observed to have the highest spread velocity of any Y. pestis clade [37] As historical plague scholarship extends beyond the bounds of Western Europe, it will be important to consider the role these lineages played, and adjust nomenclature accordingly.
Figure 4: The geographic distribution of Y. pestisBranch 2. (PLACEHOLDER)
However, a significant obstacle to understanding the global spread and virulence of past plagues lies in Y. pestis’s weak host associations and a lack of geographic structure. Similar to its parent species, Y. pseudotuberculosis[11], Y. pestis is capable of infecting a wide variety of mammalian hosts [cite?]. But while isolates of Y. pseudotuberculosis cluster by host group [39], the host structure of Y. pestis is cryptic (Figure 5). Most clades are isolated from both humans and non-human animals, although the ancient lineages 0.PRE, (Bronze Age), 0.ANT4 (Late Antiquty) and 1.PRE (Medieval/Early Modern) are exclusively associated with humans. However, this particular exception is largely due to sampling biases, as paleogenomic investigations have historically prioritized human skeletal remains over faunal remains [cite?].
Figure 5: The maximum-likelihood phylogeny of Y. pestis according to isolation host.
In addition to a cryptic host structure, the geographic patterning of Y. pestis, or lack thereof, reflects a complex dispersal history (Figure 6). Many regions have been colonized by diverse strains of Y. pestis. This diversity can be contemporaneous, such as endemic foci in the Caucausus and Western China (0.PE). Alternatively, this diversity may accrue over multiple centuries through distinct re-introductions and extinctions, as seen through historical clades in Europe (0.ANT, 1.PRE). In these examples, a relatively large amount of genetic diversity appears in a small geographic range (Figure 7). In contrast, regions such as the Americas have been colonized by a single strain of Y. pestis (1.ORI) which shows a relatively small amount of genetic diversity over a tremendously large geographic range.
This geographic complexity is unsurprising given that Y. pseudotuberculosis, the parent species of plague, also does not exhibit strong geographic structure. Outbreak strains of Y. pseudotuberculosis are particularly challenging to cluster, with non-outbreak lineages showing only slightly more geographic signal [39]. In this line of reasoning, the patterning observed in Figure 6 and 7 likely reflects the complex ecology of plague which cycles between endemic reservoirs and epidemic periods.
Figure 6: The geographic distribution of Y. pestis genomes.
Figure 7: Geographic distance vs. genetic distance. Statistical results come from a mantel test at α <= 0.05.
However, it would be amiss to not acknowledge one of the largest caveats in genomic analyses, which is the issue of sampling bias. The geographic sampling strategy of Y. pestis genomes (Figure 6) does not reflect the known distribution of modern plague [40]. The over-sampling of East Asia has been previously described by [41] and considerably drives the hypothesis that Y. pestis originated in China [17,42]. This once established hypothesis is now in contention, as the most basal strains of Y. pestis (Clades 0.PRE and 0.PE) have been isolated from all across Eurasia.
The sampling strategy of ancient DNA also does not reflect the hypothesized distribution of ancient plague. Historical genomes of Y. pestis have primarily been collected from European archaeological sites, with the most heavily sampled region being Western Europe (See Figure [map_all_branch_major?], 1.PRE).
Kat’s Notes:
- This section lacks an explicit conclusion and a transition to the next section.
Phylodynamics
Detecting Temporal Signal
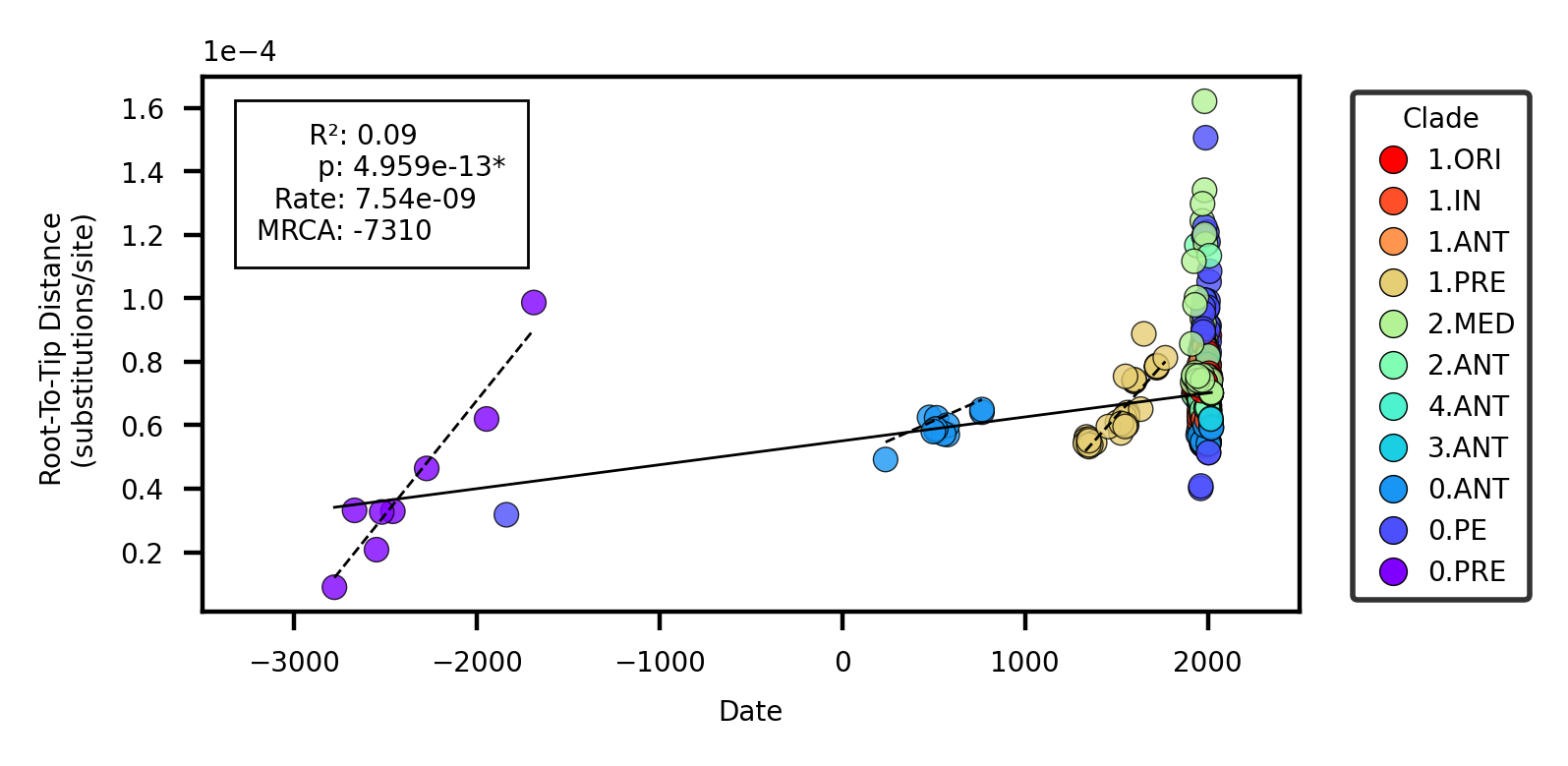
Previous work has documented substantial rate variation both between and within clades of Y. pestis[17,19]. A root-to-tip regression on sampling date for the full dataset (N=601) reproduces this finding as the coefficient of determination (R2) is extremely low at 0.09 (Figure 8). A Bayesian Evaluation of Temporal Signal (BETS) also indicates poor support for a strict clock as the coefficient of variation was consistently estimated to be greater than 1. Taken together, the root-to-tip regression and BETS analysis suggest that alternative clock models, such as the uncorrelated relaxed log normal (UCLN) model, should be preferred when accounting for the high degree of rate variation.
Figure 8: Root-to-tip regression. The solid line represents the linear model for the entire dataset. The dashed lines present linear models for clades with significant p values.
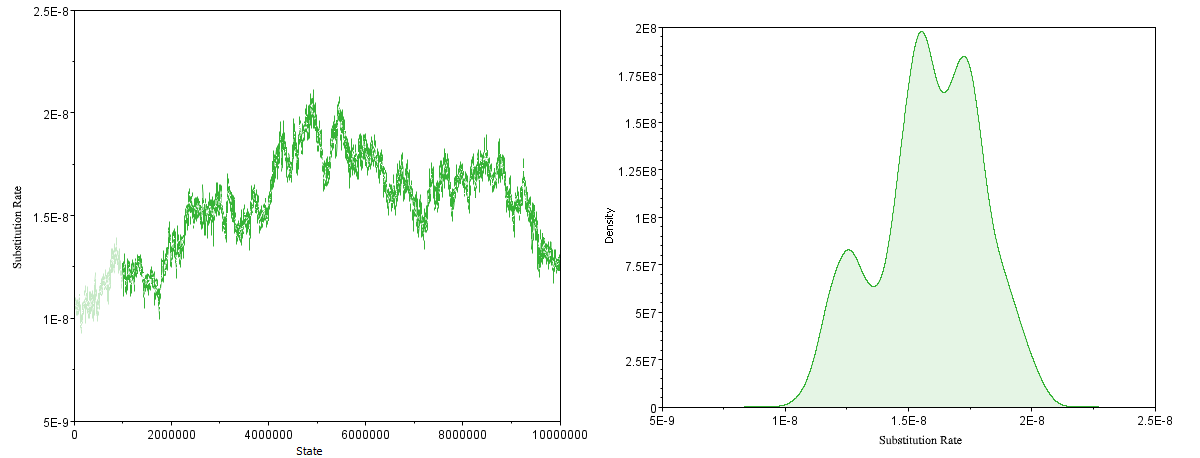
However, the BETS analysis exhibited poor sampling of the relaxed clock model parameters, even when using a fixed topology (Figure 9). This suggests there may be too much rate variation to confidently estimate key parameters such as the mean substitution rate and the time to the most recent common ancestor (tMRCA). This observation is consistent with previous analyses [18] where robust estimates of model parameters could not be estimated, thus leading to the conclusion that Y. pestis lacks temporal signal. At the same time, other studies have suggested data composition is a strong determinant of temporal signal [20] and thus we investigated alternative approaches.
Figure 9: MCMC parameter estimation of the mean substitution rate for the reduced dataset (N=191). Left: Poor mixing of the MCMC Chain, Right: The resulting multimodal estimate of the rate.
To identify patterns in rate variation that may improve the clock model, we first performed visual inspection of the root-to-tip regression residuals (Figure 8). 3/12 clades appeared to have temporal signal according to a linear model, namely the ancient clades isolated from human skeletal remains: 0.PRE (Bronze Age), 0.ANT4 (Late Antiquity), and 1.PRE (Medieval/Early Modern). Indeed, when the root-to-tip regression was performed on clades in isolation, these three clades demonstrated strong evidence of strict-clock behavior (Table 3, Figure 13). A BETS analysis by clade proved even more sensitive as temporal signal was detected in 7/12 clades (Table 1). Furthermore, for all clades with temporal signal, the relaxed clock model (UCLN) had higher support than the strict clock.
The ubiquitous support for a relaxed clock model was initially surprising, as the root-to-tip regression suggested strict clock-like behavior in several clades. However, this disparity can largely be explained by the known statistical limitations of a root-to-tip regression [43] which assumes either 1) no temporal structure, or 2) temporal structure following a linear model. Thus, a root-to-tip regression is strictly a test of the linear model, and will give no indication that other models are a better fit to the data, ie. a relaxed lognormal model. From this finding, we conclude that a root-to-tip regression is a poor statistical test of temporal signal in Y. pestis, and great caution should be taken in interpreting the associated statistics.
Kat’s Notes:
1. Could I get finalized Bayes Factors for the full dataset and clades?
2. Could I get log files for all clades, even those without temporal signal?
Table 1: Temporal signal detection and clock model selection with Bayesian Evaluation of Temporal Signal (BETS)
Clade
N
SC Bayes Factor
UCLN Bayes Factor
Clock Model
1.ORI
117
29.6*
35.7*
UCLN
1.IN
39
-3.9
-10.2
UCLN
1.ANT
4
8.9*
12.6*
UCLN
1.PRE
40
10.1*
44.1*
UCLN
2.MED
116
TBD
TBD
TBD
2.ANT
54
-20.8
-13.7
UCLN
4.ANT
11
-2.9
3.7*
UCLN
3.ANT
11
-9.6
-11.4
UCLN
0.ANT
103
-2.3
-6.5
UCLN
0.ANT4
12
5.3*
5.9*
UCLN
0.PE
83
-82.1
12.4*
UCLN
0.PRE
8
TBD
TBD
TBD
Rate Variation
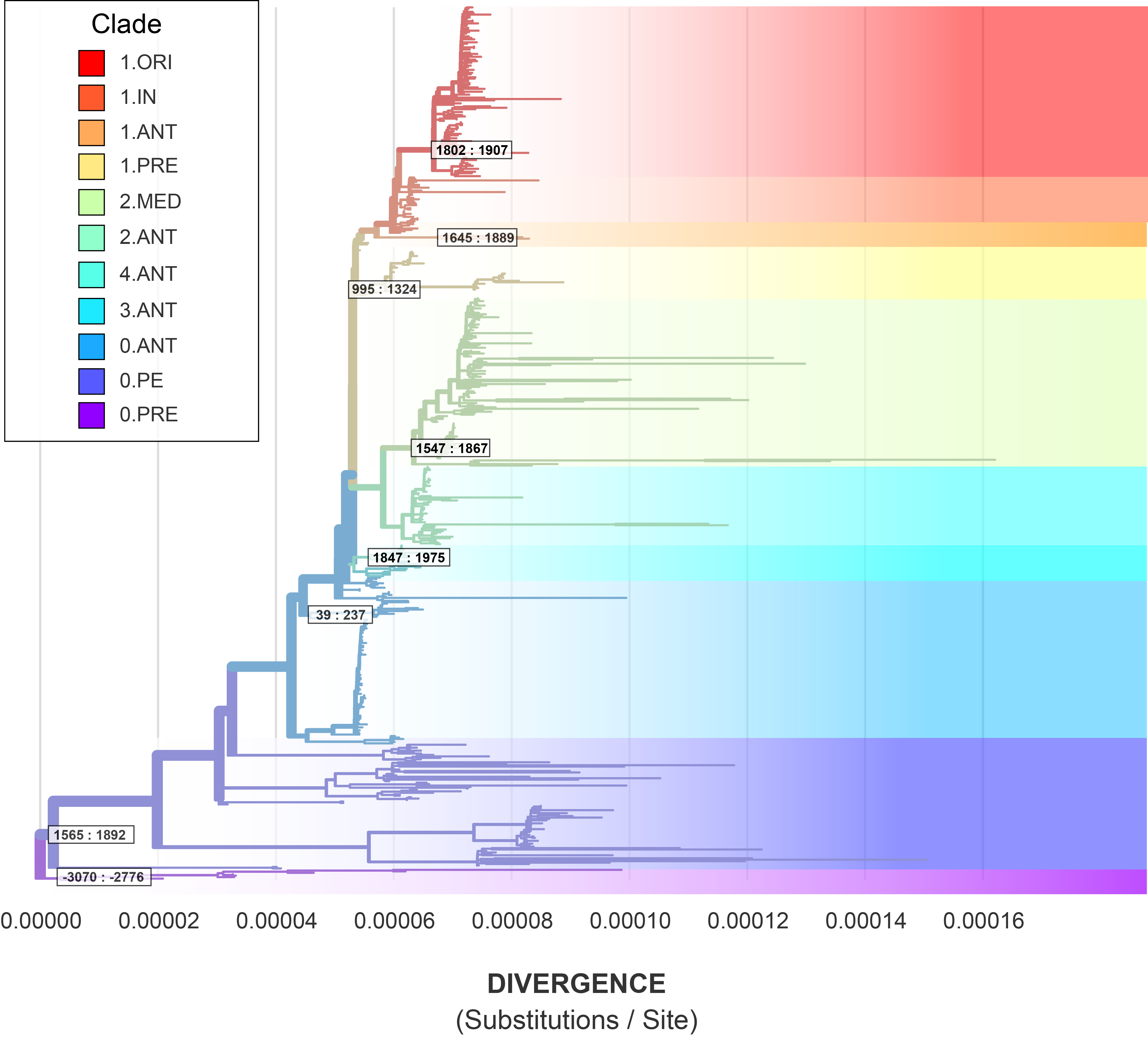
Our approach of fitting nuanced models segregated by clade reveals that the ‘true’ substitution rates of Y. pestis may be much higher than previously thought. Previous work estimated that Y. pestis has one of the slowest observed substitution rates, around 1-2 x 10-8, which is on par with the exceptionally slow-evolving Mycobacterium leprae[19]. The BETS analysis on the non-segregated data, which was highly unstable, fell within this published range with a 95% HPD between 1.16 x 10-8 and 1.95 x 10-8. However, this global rate is a considerable underestimate, as clades with detectable temporal signal ranged from 2.33 x 10-8 to 7.70 x 10-7 (Table 2, Figure 10).
Kat’s Notes:
- 0.PE appears to be an outlier, but wait for finalized logs.
- No differences with regards to rate/variation between pandemic and non-pandemic clades.
- I really want to see 1.IN, is there a progressive increase in rate along Branch 1?
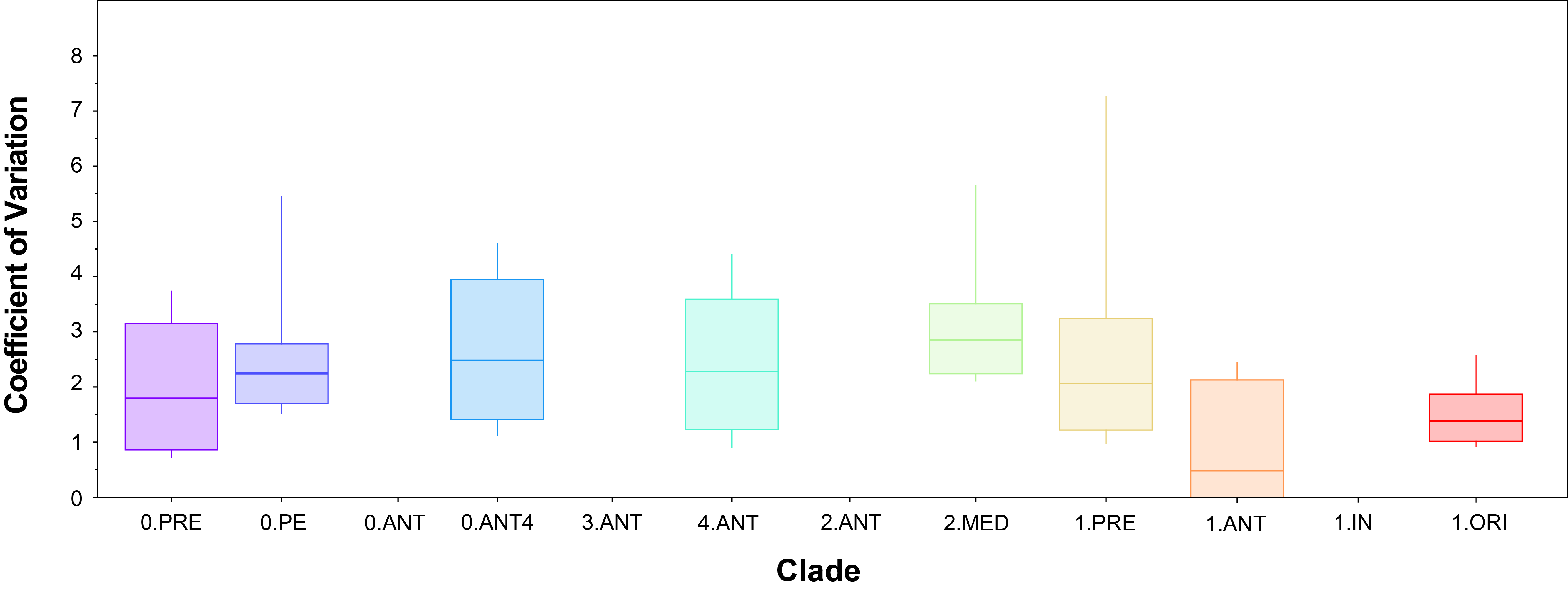
It is interesting to note that several clades have distinctly different substitution rates (ie.non-overlapping estimates of the mean substitution rate in Figure 10) and yet the relative amount of rate variation within a clade is similar (ie. overlapping coefficients of variation in Figure 11).
We hypothesize that outlier clades which are challenging to model (ex. 2.MED) have artificially decreased estimates of the mean substitution rate in past studies. This study therefore reports the substitution rate of Y. pestis to be much higher than previously thought and more comparable to bacteria such as Mycobacterium tuberulcosis.
Table 2: Estimate variation on the rate and tMRCA based on the 95% HPD.
Clade
N
Substitution Rate
Coefficient of Variation
tMRCA
All
191
1.16 x 10-8 : 1.95 x 10-8
1.35 : 2.10
-7400 : -3289
1.ORI
117
1.04 x 10-7 : 1.53 x 10-7
1.02 : 1.87
1802 : 1907
1.IN
39
–
–
–
1.ANT
4
3.61 x 10-8 : 9.63 x 10-8
0.00 : 2.12
1645 : 1889
1.PRE
40
3.69 x 10-8 : 5.90 x 10-8
1.22 : 3.24
995 : 1324
2.MED
116
1.91 x 10-7, 3.01 x 10-7
2.23 : 3.50
1547 : 1867
2.ANT
54
–
–
–
4.ANT
11
3.59 x 10-8 : 1.57 x 10-7
1.22 : 3.59
1847 : 1975
3.ANT
11
–
–
–
0.ANT
103
–
–
–
0.ANT4
12
2.33 x 10-8 : 4.68 x 10-8
1.40 : 3.94
39 : 237
0.PE
83
4.32 x 10-7 : 7.70 x 10-7
1.70 : 2.78
1565 : 1892
0.PRE
8
3.95 x 10-8 : 6.48 x 10-8
0.84 : 3.13
-3070 : -2776
Figure 10: Mean substitution rate uncertainty by clade.
Figure 11: Coefficient of variation uncertainty by clade.
Node Dating
To evaluate the dates associated with ancestral events, we annotated the maximum likelihood phylogeny with the estimated tMRCAs for clades with temporal signal (Figure 12). When re-contextualized into the global phylogeny, the node dates are topologically non-conflicting, meaning that parent nodes correctly pre-date child nodes. The sole exception is the estimated divergence of modern 0.PE which conflicts with the dates associated with the First and Second Pandemic clades. This conflict can be explained by several observations:
Clade 0.PE has the largest amount of uncertainty concerning the substitution rate.
Clade 0.PE has the greatest pairwise genetic distances, and longest root-to-tip distances to their MRCA.
The single ancient sample of 0.PE was excluded from the BEAST analysis! This was partly by mistake, as for other clades, I separated out the ancient and modern samples based on their drastically different sampling periods. But for 0.PE, there’s only one ancient sample and it got dropped.
Figure 12: The maximum likelihood phylogeny annotated with the 95% HPD on clade root date.
This drastic improvement in model performance reveals four intriguing aspects about the evolution of Y. pestis.
The first aspect is that fitting a single clock model to the global phylogeny of Y. pestis is not statistically supported. This can be observed in the relative instability of the MCMC analyses on the reduced dataset, which fails to converge in parameter space. In contrast, successfully fitting models on a clade-by-clade basis reveals that Y. pestis has more temporal signal than previously thought. The observation that different populations have evolved at drastically different rates may explain the previous finding that the apparent structure in Y. pestis is dependent on dataset composition [20].
The final finding from constructing nuanced models concerns the outlier clades for which no detectable signal could be found, namely 0.ANT, 2.ANT, 3.ANT, 2.MED, and 1.IN. The reason for this lack of signal is unclear, but one explanation may be that these Y. pestis populations are inappropriately separated based on the major branch and biovar systems. We hypothesis that alternative strategies to subdivide these populations will yield greater insight, based on the methodological improvements demonstrated in this study.
Conclusion
Fitting a single clock model to the global phylogeny of Y. pestis is not statistically supported. This can be observed in the relative instability of the MCMC analyses on the reduced dataset, which fails to converge in parameter space.
Y. pestis has much more temporal signal than previously thought.
Separating the genomic dataset by clade recovers robust temporal signal for the majority of clades.
The true substitution rates of Y. pestis are much higher than previously thought.
The mean substitution rate of all global populations (1.59E-8) is a drastic underestimate compared to the rates observed by clade which range from 3.51E-8 to 1.29E-7. The clades without temporal signal are pulling down the mean estimate. Previous work estimated that Y. pestis has one of the lowest observed substitution rates, on par with the exceptionally slow-evolving Mycobacterium leprae[20]. This study instead reports the substitution rate of Y. pestis to be much higher, and comparable to Mycobacterium tuberulcosis.
Root-to-tip regression is a poor statistical test of temporal signal compared to BETS.
The root-to-tip regression has several known limitations, namely the underlying assumption of strict clock-behavior and the non-independence of data points [43]. A BETS analysis counters these statistical violates, and is overall more sensitive given that multiple clock models can be tested. In this study, root-to-tip regression indicated temporal signal in 3/12 lineages while the BETS analysis detected signal in 7/12 lineages.
References
1.
The Stone Age Plague and Its Persistence in Eurasia
Aida Andrades Valtueña, Alissa Mittnik, Felix M Key, Wolfgang Haak, Raili Allmäe, Andrej Belinskij, Mantas Daubaras, Michal Feldman, Rimantas Jankauskas, Ivor Janković, … Johannes Krause
Early Divergent Strains of Yersinia pestis in Eurasia 5,000 Years Ago
Simon Rasmussen, Morten Erik Allentoft, Kasper Nielsen, Ludovic Orlando, Martin Sikora, Karl-Göran Sjögren, Anders Gorm Pedersen, Mikkel Schubert, Alex Van Dam, Christian Moliin Outzen Kapel, … Eske Willerslev
Population structure of the Yersinia pseudotuberculosis complex according to multilocus sequence typing
Riikka Laukkanen-Ninios, Xavier Didelot, Keith A Jolley, Giovanna Morelli, Vartul Sangal, Paula Kristo, Priscilla FM Imori, Hiroshi Fukushima, Anja Siitonen, Galina Tseneva, … Mark Achtman
Phylogeny and classification of <i>Yersinia pestis</i> through the lens of strains From the plague foci of Commonwealth of Independent States
Vladimir V Kutyrev, Galina A Eroshenko, Vladimir L Motin, Nikita Y Nosov, Jaroslav M Krasnov, Lyubov M Kukleva, Konstantin A Nikiforov, Zhanna V Al’khova, Eugene G Oglodin, Natalia P Guseva
The EnteroBase user's guide, with case studies on <i>Salmonella</i> transmissions, <i>Yersinia pestis</i> phylogeny, and <i>Escherichia</i> core genomic diversity
Zhemin Zhou, Nabil-Fareed Alikhan, Khaled Mohamed, Yulei Fan, Mark Achtman
<i>Yersinia pestis</i> and the Plague of Justinian 541–543 AD: a genomic analysis
David M Wagner, Jennifer Klunk, Michaela Harbeck, Alison Devault, Nicholas Waglechner, Jason W Sahl, Jacob Enk, Dawn N Birdsell, Melanie Kuch, Candice Lumibao, … Hendrik Poinar
Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes
Maria A Spyrou, Marcel Keller, Rezeda I Tukhbatova, Christiana L Scheib, Elizabeth A Nelson, Aida Andrades Valtueña, Gunnar U Neumann, Don Walker, Amelie Alterauge, Niamh Carty, … Johannes Krause
Reproducible, portable, and efficient ancient genome reconstruction with nf-core/eager
James AFellows Yates, Thiseas C Lamnidis, Maxime Borry, Aida Andrades Valtueña, Zandra Fagernäs, Stephen Clayton, Maxime U Garcia, Judith Neukamm, Alexander Peltzer
Ancient <i>Yersinia pestis</i> genomes from across Western Europe reveal early diversification during the First Pandemic (541–750)
Marcel Keller, Maria A Spyrou, Christiana L Scheib, Gunnar U Neumann, Andreas Kröpelin, Brigitte Haas-Gebhard, Bernd Päffgen, Jochen Haberstroh, Albert Ribera i Lacomba, Claude Raynaud, … Johannes Krause
Evolution and circulation of Yersinia pestis in the Northern Caspian and Northern Aral Sea regions in the 20th-21st centuries
Galina A Eroshenko, Nikolay V Popov, Zhanna V Al’khova, Lyubov M Kukleva, Alina N Balykova, Nadezhda S Chervyakova, Ekaterina A Naryshkina, Vladimir V Kutyrev
Genomic Insights into a Sustained National Outbreak of Yersinia pseudotuberculosis
Deborah A Williamson, Sarah L Baines, Glen P Carter, Anders Gonçalves da Silva, Xiaoyun Ren, Jill Sherwood, Muriel Dufour, Mark B Schultz, Nigel P French, Torsten Seemann, … Benjamin P Howden
Historical and genomic data reveal the influencing factors on global transmission velocity of plague during the Third Pandemic
Lei Xu, Leif C Stige, Herwig Leirs, Simon Neerinckx, Kenneth L Gage, Ruifu Yang, Qiyong Liu, Barbara Bramanti, Katharine R Dean, Hui Tang, … Zhibin Zhang
Historical <i>Y. pestis</i> Genomes Reveal the European Black Death as the Source of Ancient and Modern Plague Pandemics
Maria A Spyrou, Rezeda I Tukhbatova, Michal Feldman, Joanna Drath, Sacha Kacki, Julia Beltrán de Heredia, Susanne Arnold, Airat G Sitdikov, Dominique Castex, Joachim Wahl, … Johannes Krause
 0000-0001-6862-7756
·
0000-0001-6862-7756
·  ktmeaton
ktmeaton